
Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма


Рубрика: Эхография в акушерстве
УЗ сканер Samsung HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Синдром Тричера Коллинза (СТК, Treacher Collins syndrome) – это врожденное, наследственно обусловленное нарушение развития производных первой глоточной дуги, которое характеризуется специфическими черепно-лицевыми проявлениями: двусторонней симметричной отонижнечелюстной дисплазией с гипоплазией скуловых костей.
Синонимы: синдром Франческетти, синдром Тричера Коллинза–Франческетти, синдром Франческетти–Цвалена–Клейна, челюстно-лицевой дизостоз.
Отличительные признаки СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, опущенные уголки глаз, колобома нижнего века, пороки развития наружного уха [1, 2].
Хотя первым болезнь описал Аллен Томсон еще в 1846 г., синдром обычно называют именем врача Тричера Коллинза, который в 1900 г. описал двух больных с похожими симптомами. Не верно писать этот синдром через дефис, так как Тричер – имя доктора Коллинза. Уже в 40-х годах прошлого века Адольф Франческетти и Давид Клейн дали подробную характеристику болезни и назвали ее челюстно-лицевым дизостозом [3]. В некоторых странах Европы этот синдром называют синдромом Франческетти или синдромом Тричера Коллинза–Франческетти [4, 5].
Популяционная частота СТК оценивается как 1:50 000 живорожденных [1, 2], однако некоторые авторы называют более частую встречаемость этого синдрома: 1:10 000 [6]. Больные легко узнаваемы, их можно нередко встретить на улицах, увидеть в социальных сетях и, иногда, на телеэкранах. В 2017 г. вышла кинокартина режиссера Стивена Чбоски с Джулией Робертс в главной роли, которая называется «Чудо», где рассказана история мальчика Огги Пулмана с синдромом Тричера Коллинза и прекрасно продемонстрирована вся сложность социальной адаптации таких детей.
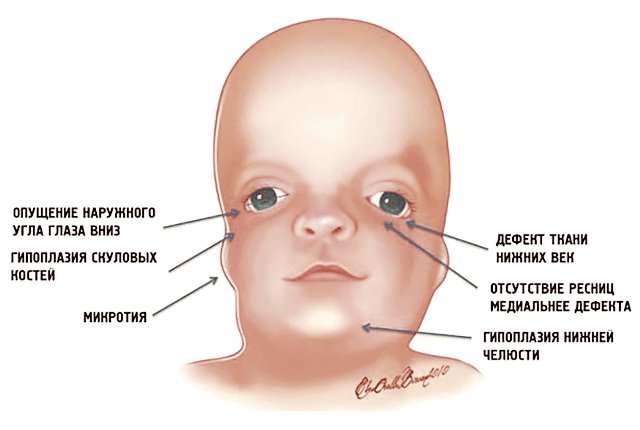
Рис. 1. Схема специфических признаков лицевых дизморфий при синдроме Тричера Коллинза.
Наиболее частые симптомы и фенотипические проявления СТК
У людей с СТК отмечается характерный лицевой дизморфизм (рис. 1) с двусторонней симметричной гипоплазией скуловых костей (95%), характерна гипоплазия инфраорбитального края глазницы (80%) с формированием антимонголоидного разреза глаз (89%) и гипоплазией нижней челюсти (78%), что приводит к аномалии прикуса [1–6], также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан [7], колобома (расщелина) нижних век между внешней и средней третью (69%), сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной воз можности открытия рта различной степени тяжести [1].
Часто отмечается аномалия наружного уха, например микротия или анотия (77%), атрезия наружного слухового прохода и аномалии развития слуховых костей (60%), что приводит к кондуктивной тугоухости [1–8]. Снижение зрения, вплоть до полной его потери, встречается в 37% случаев. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина (28%).
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК [1, 2]. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием [8]. Из частых признаков описан чрезмерный рост волос на щеках [2, 8, 9].
Этиология синдрома Тричера Коллинза
На сегодняшний день описано три типа СТК. До 93% всех случаев – это синдром 1-го типа [10]. СТК 1-го типа связан с мутациями гена TCOF1, который расположен в сегменте 5q32 – q33. Тип наследования аутосомно-доминантный [2] с 90% пенетрантностью и переменной экспрессивностью (проявляемостью), даже у пациентов в пределах одной семьи. Известны наблюдения детей с выраженными клиническими проявлениями синдрома в одной семье, тогда как у одного из их родителей была обнаружена та же мутация без выраженных клинических проявлений болезни [2, 4–6]. Около 60% случаев СТК не наследуются от больных родителей, а являются новыми мутациями (de novo).
Также описаны 2-й и 3-й типы СТК. Второй тип вызван мутацией гена POLR1D на хромосоме 13q12, 3-й тип – мутацией гена POLR1C на хромосоме 6p21. Нужно отметить, что клинически все три типа не отличаются друг от друга, несмотря на то что мутации затрагивают разные гены, на разных хромосомах [2] и тип наследования может быть и аутосомно-рецессивным [11].
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии [12]. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода [14, 15]. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику [16]. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной [16, 17].
Дифференциальная диагностика СТК должна включать некоторые генетические синдромы с преимущественным поражением лицевых структур [17]:
- Синдром Гольденхара. Изменения лица при синдроме Гольденхара почти всегда односторонние, асимметричные, включают в себя колобому верхнего, а не нижнего века, а также эпибульбарные дермоиды, преаурикулярные привески. При синдроме Гольденхара могут встречаться аномалии позвоночника и пороки сердца.
- Синдром Нагера. Фенотипически похож на СТК, однако для него характерны преаксиальные (со стороны большого пальца кисти) дефекты верхней конечности – редукционные пороки верхних конечностей (в диапазоне от гипоплазии до аплазии большого пальца с или без вовлечения лучевой кости).
- Синдром Миллера, известный как постаксиальный акрофациальный дизостоз. Характеризуется микрогнатией, расщелиной губы, различными аномалиями позвонков и сколиозом. Типичными признаками являются постаксиальные (со стороны мизинца кисти) пороки верхней конечности либо только мизинца.
- Синдром Пьера Робена характеризуется изолированной гипоплазией нижней челюсти, глоссоптозом, расщелиной нёба.
Следует подчеркнуть, что аномалии конечностей не свойственны для СТК и для синдрома Пьера Робена, и, если они присутствуют, следует больше думать о синдромах Миллера или Нагера.
Профилактика и лечение СТК
Генетическое консультирование семей с больным ребенком/плодом осложняется вариабельной проявляемостью заболевания и должно осуществляться мультидисциплинарной группой специалистов по пренатальной диагностике с обязательным выяснением этиологии возникновения заболевания в конкретной ситуации (семейная форма либо мутация de novo). При наличии у родителя признаков СКР единственным эффективным методом профилактики заболевания следует назвать применение методик экстракорпорального оплодотворения с предимплантационной диагностикой с целью переноса здоровых эмбрионов, либо применение донорских ооцитов или сперматозоидов.
При продолжающейся беременности послеродовое ведение требует междисциплинарного подхода (акушер, неонатолог, хирург, анестезиолог и генетик); и из-за возможных острых проблем с дыханием роды должны планироваться в специализированных перинатальных центрах. Лечение больных с СТК многопрофильное. В случае возникновения постнатального респираторного дистресс-синдрома необходимо применение трахеостомии, неинвазивной вентиляции и дистракции нижней челюсти. Челюстно-лицевая и пластическая хирургия позволяет устранить гипоплазию мягких тканей (коррекция овала лица с помощью липоскульптуры), гипоплазию костной ткани (хирургическая дистракция кости, костные трансплантаты), колобому век и расщелину нёба (хирургическое восстановление). Для устранения аномалий среднего уха (функциональная хирургия) и наружного уха (реконструкция ушных раковин) требуется участие специалиста в области ЛОР-хирургии. Коррекция нарушения слуха должна осуществляться на ранней стадии (слуховые аппараты и функциональная хирургия), что способствует нормальному развитию ребенка.
При надлежащем лечении прогноз для легких форм заболевания является благоприятным. Для тяжелых форм заболевания с выраженными клиническими проявлениями прогноз неблагоприятный не только для здоровья, но и для жизни.
Описание случая синдрома Тричера Коллинза
В медико-генетическом отделении (МГО) Московского областного НИИ акушерства и гинекологии для консультации по прогнозу потомства и возможностях обследования обратилась пациентка 25 лет со сроком беременности 8 нед. Данная беременность вторая. Брак не родственный. Муж здоров, производственных вредностей супруги не имеют. Первая беременность закончилась преждевременными родами в сроке 36 нед. Родилась девочка с массой тела 1990 г, ростом 51 см, с оценкой по шкале Апгар 7/7 баллов. При осмотре ребенка генетиком выявлены особенности фенотипа, характерные для СТК: гипоплазия скуловых костей, антимонголоидный разрез глаз, гипоплазия нижней челюсти, двусторонняя микротия с атрезией слуховых проходов. Методом автоматического прямого секвенирования был проведен поиск мутаций в гене TCOF1. Выявлен патогенный вариант c.3946_3947 delGA в гетерозиготном состоянии. Ребенку выставлен клинический диагноз: синдром Тричера Коллинза. Тяжесть состояния ребенка усугубилась врожденной пневмонией, церебральной ишемией II степени, недоношенностью, анемией тяжелой степени. Ребенок был переведен в отделение реанимации, умер в 1,5 мес. При консультировании ребенка генетиком риск повторного рождения больного ребенка в семье расценен как низкий, так как данная мутация расценена генетиком как мутация de novo. Дана рекомендация о пренатальной диагностике и кариотипировании плода при следующей беременности без указания на необходимость специфической диагностики СТК. Пациентка самостоятельно обратилась для обследования в медико-генетический научный центр (МГНЦ). В образце ее ДНК методом прямого автоматического секвенирования была найдена патогенная мутация в гене TCOF1 в гетерозиготном состоянии. Таким образом, у пациентки тоже имеется СТК и риск рождения у нее больных детей будет высоким – 50%. При предыдущем осмотре генетиком ее фенотип был не изучен и не оценен в полном объеме. При внимательном осмотре пациентки найдены мягкие, но классические признаки СТК: опущенные уголки глаз, колобомы нижнего века, рост волос на лице, гипоплазия мягких тканей в области скуловых дуг. При сборе анамнеза выяснено, что пациентка страдает двусторонней тугоухостью. С учетом аутосомно-доминантного типа наследования СТК, известного картированного патологического гена было рекомендовано проведение инвазивной пренатальной диагностики с прицельным поиском известной мутации и экспертное ультразвуковое исследование в 12–13 нед беременности.
При ультразвуковом исследовании выявлены множественные особенности лицевого фенотипа у плода: микрогнатия (рис. 2–4), треугольная форма лица (рис. 5), опущенные книзу глазницы и гипоплазия скуловых дуг (рис. 6, 7), аномальная форма и положение ушей (рис. 5, 7).
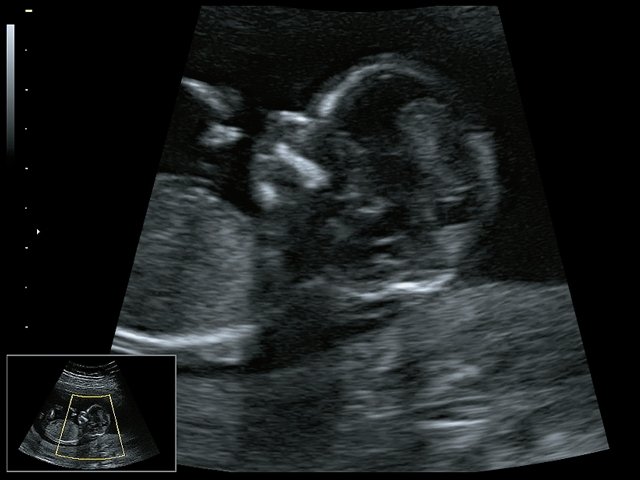
Рис. 2. Микрогнатия - сагиттальный скан в 2D, беременность 13 нед.
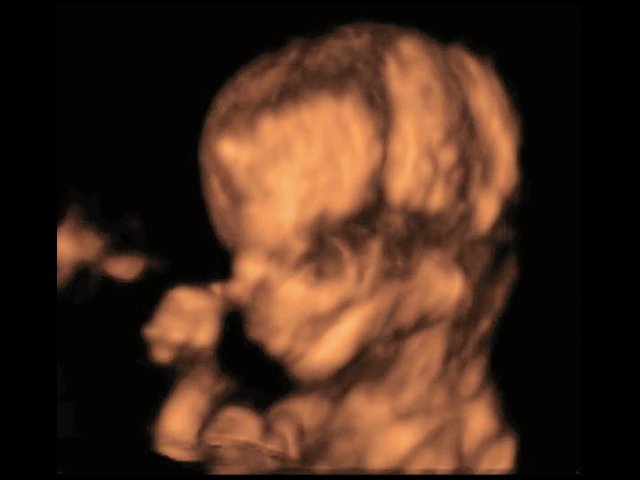
Рис. 3. Микрогнатия - 3D-реконструкция лица плода при СТК.
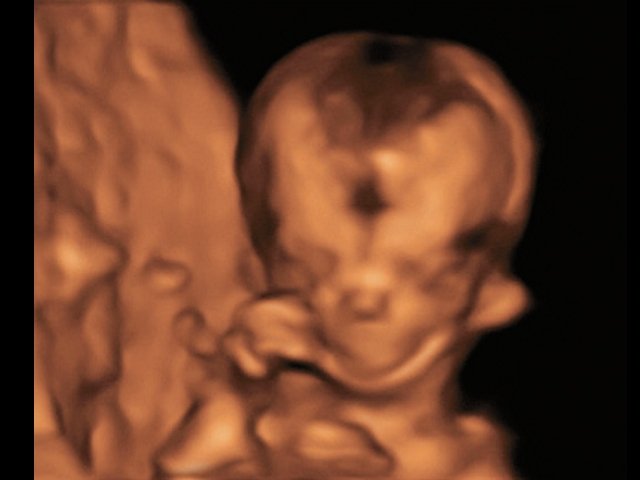
Рис. 4. Микрогнатия - 3D-реконструкция лица плода при синдроме Тричера Коллинза.
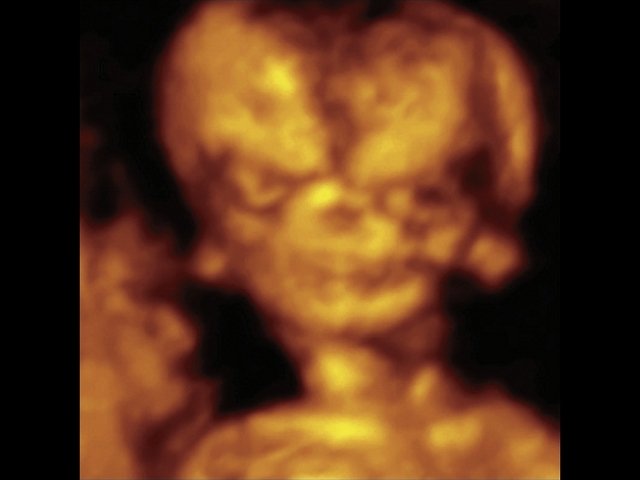
Рис. 5. Треугольная форма лица плода при синдроме Тричера Коллинза, аномальная форма и положение ушей в 3D.
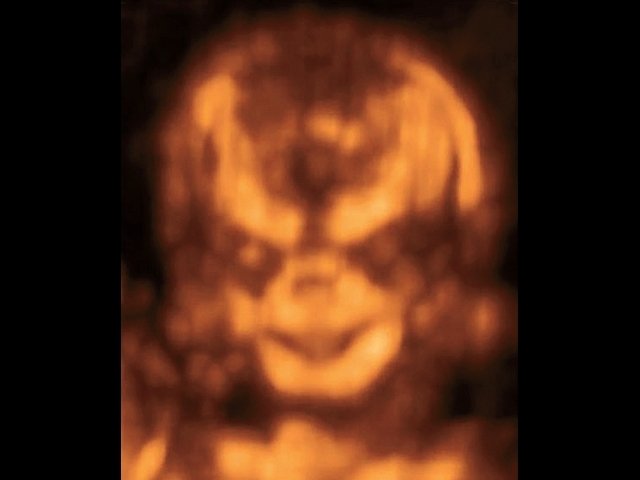
Рис. 6. 3D-сканирование лица плода с синдромом Тричера Коллинза. Опущенные книзу глазницы, гипоплазия скуловых костей.
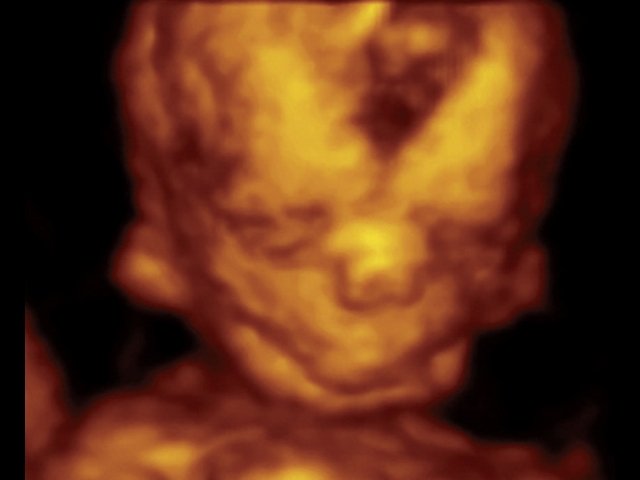
Рис. 7. Фенотип лица плода с синдромом Тричера Коллинза.
В МГНЦ по результату исследования ДНК плода в образцах плодного материала, полученного при проведении аспирации ворсин хориона, методом автоматического секвенирования у плода обнаружена патогенная мутация в гене TCOF1 (с. 3946- 3947 del GA) в гетерозиготном состоянии.
Выставлен клинический диагноз: беременность 13 нед. Синдром Тричера Коллинза у плода. Отягощенный генетический анамнез. Проведены медико-генетическое консультирование и пренатальный консилиум, прогноз для здоровья будущего ребенка определен как условно неблагоприятный. Семья приняла решение о досрочном прекращении беременности.
По результату повторного консультирования в МГО по вопросу репродуктивного поведения и прогноза потомства семья приняла решение о планировании следующей беременности с применением методик вспомогательных репродуктивных технологий с предимплантационной генетической диагностикой.
Выводы
- Оптимальным периодом для ультразвукового пренатального выявления новых случаев СТК (мутация de novo) является обследование во II триместре (в сроки второго скринингового обследования в 19–22 нед беременности), а для семейно-положительных случаев – период первого скринингового обследования (12–14 нед беременности).
- 3D/4D-изображения более наглядно и очевидно демонстрируют особенности изменения лицевого фенотипа при некоторых генетических синдромах, обеспечивая более понятную визуализацию признаков как для специалистов, так и для родителей.
- Сонографические находки при подозрении на СТК чрезвычайно важны, чтобы добавить специфическое обследование (секвенирование) в обычную панель исследования материала плодного происхождения, подтверждая диагноз.
- Учитывая варианты различной экспрессивности генов, необходимы тщательнейший осмотр пациентов (как матери, так и отца), сбор семейного анамнеза для постановки окончательного диагноза и определения риска повтора заболевания в семье и формулировки специфических, конкретных мер профилактики генетической патологии.
Литература
- Кеннет Л. Джонс Наследственные синдромы по Д. Смиту М.: Практика, 2011: 296–297.
- https://omim.org/entry/154500?search=omim%20154500&highlight=154500%20omim
- Franceschetti A., Klein D. Mandibulo-facial dysostosis: new hereditary syndrome // Acta Ophthal. 1949; 27: 143–224.
- Bowman M., Oldridge M., Archer C. et al. Gross deletions in TCOF1 are a cause of Treacher-Collins-Franceschetti syndrome // Eur J Hum Genet. 2012; 20: 769–777.
- Edery P., Manach Y., Le Merrer M. et al. Apparent genetic homogeneity of the Treacher Collins-Franceschetti syndrome // Am J Med Genet. 1994; 52: 174–177.
- Trainor P.A., Dixon J., Dixon M.J. Treacher Collins syndrome: etiology, pathogenesis and prevention // Eur J Hum Genet. 2009; 17: 275–283.
- Beryl Benacerraf. Ultrasound of Fetal Syndromes. Elsevier, 2008: 184–186.
- Vincent M., Genevieve D., Ostertag A. et al. 44 others. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients // Genet Med. 2016; 18: 49–56.
- Dixon M.J. Treacher Collins syndrome // Hum Mol Genet. 1996; 5 Spec No: 1391–1396.
- Teber O.A., Gillessen-Kaesbach G., Fischer S. et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation // Eur J Hum Genet. 2004; 12: 879–890.
- Lowry R.B., Morgan K., Holmes T.M. et al. Mandibulofacial dysostosis in Hutterite sibs: a possible recessive trait // Am J Med Genet. 1985; 22: 501–512.
- Edwards S.J., Fowlie A., Cust M.P. et al. Prenatal diagnosis in Treacher Collins syndrome using combined linkage analysis and ultrasound imaging // J Med Genet. 1996; 33: 603–606.
- Ahmed, Ahmed Zakaria, Mahy Mohsen // https://sonoworld.com/TheFetus/page.aspx?id=3819 Treacher Collins syndrome.
- Nicolaides K.H., Johansson D., Donnani D., Rodeck C.H. Prenatal diagnosis of mandibulofacial dysostosis // Prenat Diagn. 1984; 4: 201–205.
- Cohen J., Ghezi F., Goncalves I. et al. Prenatal Sonographic diagnosis of Teacher Collins syndrome: a case and review of literature // Am J Perinatol. 1995; 12: 416–419.
- Tanaka Y, Kanenishi K., Tanaka H. et al. Antenatal three-dimensional sonographic features of Treacher Collins syndrome // Ultrasound Obstet Gynecol. 2002; 19: 414–415.
- Grochal F., Dankovcik R. et al. Treacher Collins syndrome – role of 3D/4D ultrasound in the assessment of fetal facial dysmorphism // https://sonoworld.com/TheFetus/page.aspx?id=3491
УЗ сканер Samsung HS50
Доступная эффективность. Универсальный ультразвуковой сканер, компактный дизайн и инновационные возможности.
Публикации по теме
- Случай острого развития синдрома анемии-полицитемии (TAPS) у монохориальной диамниотической двойни с синдромом селективной задержки роста плода (sIUGR) - Терегулова Л.Е.
- Опухоль пуповины: ультразвуковая диагностика в I триместре - Волков А.Е.
- Случай ранней пренатальной диагностики синдрома Тричера Коллинза (Treacher Collins syndrome, OMIM: 154500) 1-й тип, семейная форма - Андреева Е.Н.
- Современные подходы к изучению плодового фенотипа в эру генетического ультразвука - Андреева Е.Н.
- Эхогистеросальпингография с применением контрастного препарата «Соновью» - Федоткина Е.П.